Sentieon开发的Hap-eval准确率评估工具在设计之初就考虑到了复杂以及重复的基因组区域,采用了基于单倍型拼接序列的矩阵比较模式,兼容包括PacBio和ONT在内的主流三代长读长测序数据。另外值得一提的是,Hap-eval基于python所写,运行效率非常高,速度快,非常适用于大规模分析场景。
开源地址:
https://github.com/Sentieon/hap-eval
工具介绍:
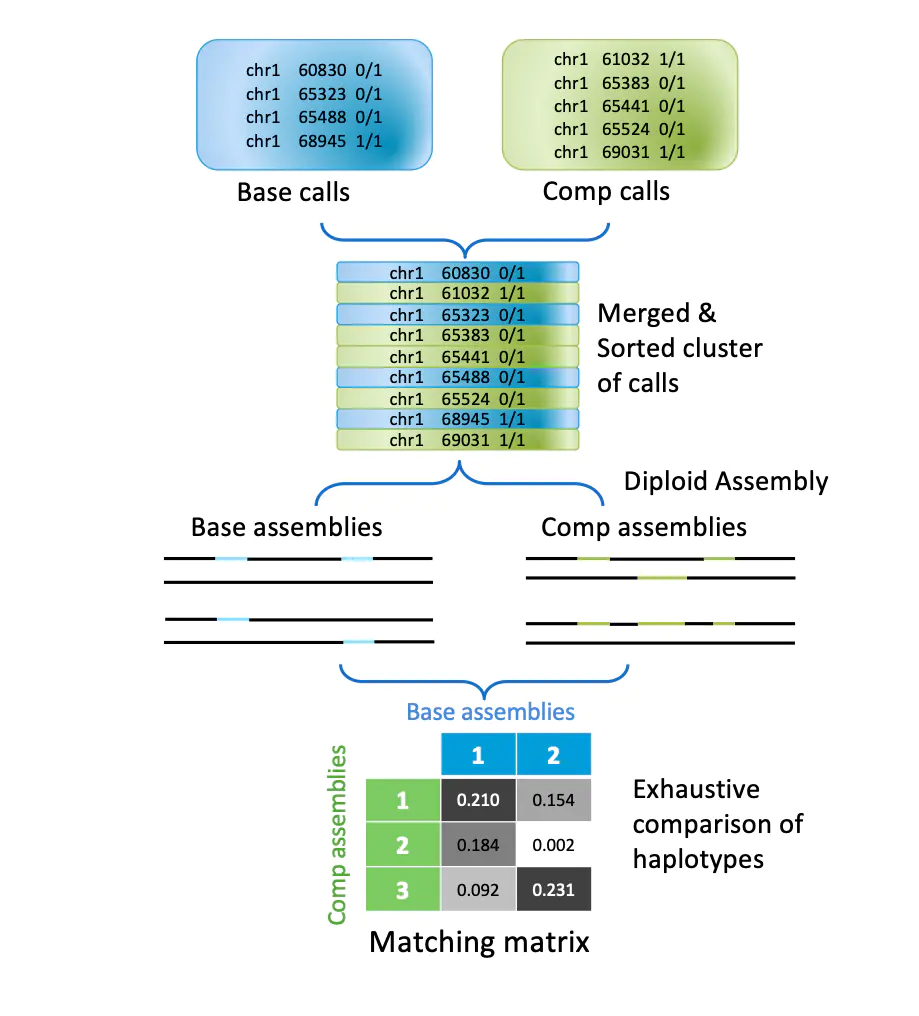
Sentieon的研发团队开发了SV评估软件 Hap-eval。Hap-eval基于单倍型 (haplotype) 对两组SV结果进行比较,首先会将比较区块内的SV拼接成单倍型序列,如果SV的结果中有定相信息,在这一步也可以被利用;然后这些单倍型序列被用来建立一个矩阵,进行结果判断。
安装方法:
git clone --recurse-submodules https://github.com/Sentieon/hap-eval.gitpip install ./hap-eval
使用方法:
usage: hap_eval [-h] -r FASTA -b VCF -c VCF [-i BED] [-t INT] [--base_out VCF] [--comp_out VCF] [--maxdist INT] [--minsize INT] [--maxdiff FLOAT] [--metric STR]optional arguments: -h, --help show this help message and exit -r FASTA, --reference FASTA Reference file -b VCF, --base VCF Baseline vcf file -c VCF, --comp VCF Comparison vcf file -i BED, --interval BED Evaluation region file -t INT, --thread_count INT Number of threads --base_out VCF Annotated baseline vcf file --comp_out VCF Annotated comparison vcf file --maxdist INT Maximum distance to cluster variants (default: 1000) --minsize INT Minimum size of variants to consider (default: 50) --maxdiff FLOAT Haplotype difference theshold (default: 0.2) --metric STR Distance metric (default: Levenshtein)
Hap-Eval 是一款新发布软件,功能在持续完善开发中。如果 遇到不能Hap-Eval不能适配的 VCF文件,可通过github或sentieon@insvast.com及时反馈。